
Under most circumstances, fungi coexist with trees or act as saprotrophs responsible for carbon and nitrogen cycling in forest systems. The order Hymenochaetales is dominated by wood decay fungi and belongs to Agaricomycetes in Basidiomycota. Most species in this order are saprotrophic or weakly pathogenic, but a few of these species exhibit strong pathogenicity. In various parts of the world, there has been an emergence of tree disease outbreaks, including brown root rot caused by Phellinus noxius and laminated root rot caused by Phellinus sulphurascens. Despite the ecological importance of these fungi and the impact of the diseases that they cause, little is known about their evolution and transmission patterns.
During the last 20 years, brown root rot disease has become a serious threat to a variety of agricultural, ornamental, landscape, and forest trees in Taiwan; the Ryukyu Islands and the Ogasawara Islands of Japan; Hong Kong; and Macao. The pathogen P. noxius has an extremely wide host range that spans more than 200 broad-leaved and coniferous tree species (in at least 59 families), such as longan, litchi, camphor, banyan, and pine trees. The corresponding disease is widespread in tropical and subtropical areas in Southeast Asia, East Asia, Oceania, Africa, Central America and the Caribbean. This geographical distribution appears to be related to the growth temperature range of P. noxius, which is 10-12°C to 36°C, with optimum growth at 30°C. Although P. noxius is recognized as an emerging destructive pathogen that causes tree death, its genome, evolution and global population genetics are poorly understood.
In a joint international effort led by Dr. Isheng J. Tsai from the Biodiversity Research Center, Academia Sinica, and Dr. Chia-Lin Chung from the Department of Plant Pathology and Microbiology of National Taiwan University (NTU), who collaborated with the Japan Forestry and Forest Products Research Institute, University of Miyazaki, Japan, as well as Taiwan Agricultural Research Institute and NTU’s Department of Bio-industrial Mechatronics Engineering, a reference genome of P. noxius and high-quality assemblies of three closely related Hymenochaetales species (the wood-decomposing fungus P. lamaensis, the laminated root rot fungus P. sulphurascens and the trunk pathogen Porodaedalea pini) were released.
Long-read sequencing was used to sequence the ~31 Mb genome of P. noxius to a high level of completion; notably, telomere-to-telomere chromosome sequences were obtained. Comparative genomics analyses of Hymenochaetales genomes identified many gene families of lignin-degrading enzymes, reflecting the examined organisms’ capacity to act as white rot fungi. The P. noxius genome contained highly expanded 1,3-beta-glucan synthase genes, which may account for its fast-growing nature. A total of 4.23% of the P. noxius proteome was annotated as carbohydrate-active enzymes (CAZymes); this percentage was higher than those observed for other Hymenochaetales species. The comprehensive repertoire of CAZymes encoded in the P. noxius genome suggests this fungus’s capability to infect a wide range of hosts.
To further understand the biology and regional dissemination of P. noxius, the research team sequenced the genomes of 60 isolates originating from diseased trees across Taiwan and offshore Japanese islands from 2007-2014. Population genomics analyses suggested that P. noxius is a hypervariable species for which both monokaryotic and heterokaryotic states of mycelia are prevalent in nature. Investigations into mating type loci and genome-wide heterozygosity indicated that this organism’s genetic hyperdiversity can be attributed to its bipolar heterothallic reproductive system and heterokaryotic nature. Evidence from whole-genome sequencing and simple sequence repeat (SSR) markers suggested that P. noxius may disseminate over short distances via root-to-root contact between host plants and that genetically variable basidiospores are likely responsible for long-distance dispersal. Moreover, long-distance dispersal via the transport of infected plants/stumps or colonized wood debris by human activities cannot be excluded. In addition, it was revealed that P. noxius from the Ryukyu Islands may have originated in Taiwan and that P. noxius in the Ogasawara Islands (located 1,210 km from the Ryukyu Islands) may be derived from a genetically distinct gene pool and could have undergone divergent evolution. The different evolutionary histories of these two P. noxius populations could reflect different times of introduction, different environments, and human interference in the Taiwan-Ryukyu and Ogasawara regions.
The genetic hyperdiversity of P. noxius suggests that this pathogen may be highly adaptable to different environments and stresses. Much more attention must be devoted to how global warming and the increasing frequency of extreme weather events will exacerbate disease severity and allow a fast-growing pathogen such as P. noxius to colonize warm temperate zones.

Figure 1. Life stages of P. noxius and a comparative genomic analysis of Hymenochaetales species. (a) P. noxius mainly infects the roots of the host tree, with infections sometimes accompanied by gradual expansion of the mycelial mat to the basal stem. The mycelial mat has a young, cream-colored leading front and an aged brown section. (b) In trees in an advanced stage of decay, the hyphae form a network of brown zone lines that permeate the soft, white wood tissue. (c, d) Basidiocarps are perennial and can be resupinate (c) or grow into a sessile, bracket-like conk with a broad basal attachment (d). The distinctive grayish-brown surface is the hymenial layer with irregular polygonal pores that contains four-spored basidia and ellipsoid and hyaline basidiospores but no hymenial setae. (e) The phylogeny of the four Phellinus species sequenced in this study. P. noxius and P. lamaensis are more closely related and have smaller genomes (~31 Mb); P. pini and Fomitiporia mediterranea have larger genomes (53-63 Mb).
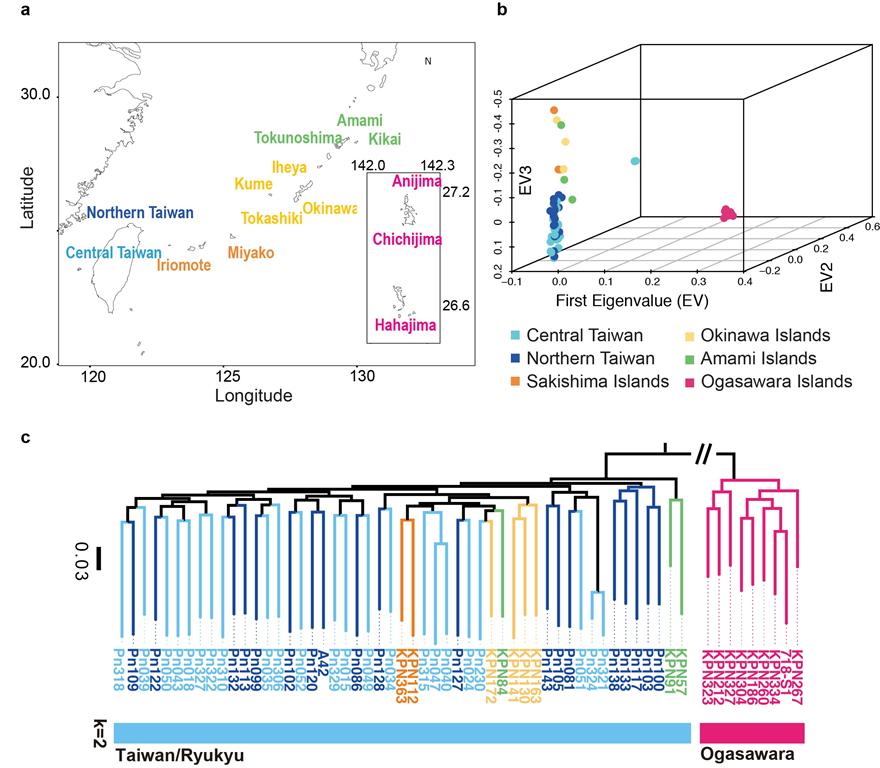
Figure 2. Map of Taiwan and offshore Japanese islands that shows the origins of the 60 P. noxius isolates sampled from 2007-2014. P. noxius isolates from Taiwan and the Ryukyu Islands belong to the same lineage, whereas isolates from the Ogasawara Islands (1,210 km apart) are genetically distinct.
Source of the materials
Paper link
References
- Chia-Lin Chung,* Tracy J. Lee, Mitsuteru Akiba, Hsin-Han Lee, Tzu-Hao Kuo, Dang Liu, Huei-Mien Ke, Toshiro Yokoi, Marylette B. Roa, Meiyeh J. Lu, Ya-Yun Chang, Pao-Jen Ann, Jyh-Nong Tsai, Chien-Yu Chen, Shean-Shong Tzean, Yuko Ota, Tsutomu Hattori, Norio Sahashi, Ruey-Fen Liou, Taisei Kikuchi,* and Isheng Jason Tsai* (2017). Comparative and population genomic landscape of Phellinus noxius: A hypervariable fungus causing root rot in trees. Molecular Ecology, 26(22), 6301-6316. DOI:10.1111/mec.14359.
- Chia-Lin Chung, Shun-Yuan Huang, Yu-Ching Huang, Shean-Shong Tzean, Pao-Jen Ann, Jyh-Nong Tsai, Chin-Cheng Yang, Hsin-Han Lee, Tzu-Wei Huang, Hsin-Yu Huang, Tun-Tschu Chang, Hui-Lin Lee, and Ruey-Fen Liou* (2015). The genetic structure of Phellinus noxius and dissemination pattern of brown root rot disease in Taiwan. PLoS ONE, 10(10), e0139445. DOI:10.1371/journal.pone.0139445.